
The dynamic role of molecules to support the life is documented since the initial stages of biological research. Proteomics is one of the most significant methodologies to comprehend the gene function although, it is much more complex compared with genomic. Fluctuations in gene expression level can be determined by analysis of transcriptome or proteome to discriminate between two biological states of the cell. Proteomics study is crucial for early disease diagnosis, prognosis and to monitor the disease development. Furthermore, it also has a vital role in drug development as target molecules. Proteomics is the characterization of proteome, including expression, structure, functions, interactions and modifications of proteins at any stage. The proteome also fluctuates from time to time, cell to cell and in response to external stimuli. Proteomics in eukaryotic cells is complex due to post-translational modifications, which arise at different sites by numerous ways.
The conventional methodology for protein analysis includes protein extraction, purification and structural studies. Cells or tissue are processed by various physical (sonication) and chemical (detergents) techniques for the extraction of total protein. Based upon physiochemical nature of polypeptides, the protein of interest can be separated out by different chromatographic techniques. Various methods including X-ray crystallography, NMR and MALDI-TOF are extensively used for structural elucidation and functional characterization of proteins. Nowadays, High-throughput techniques including total proteome analysis and MALDI-TOF are employed to study protein network biology.

Protein Screening and Identification
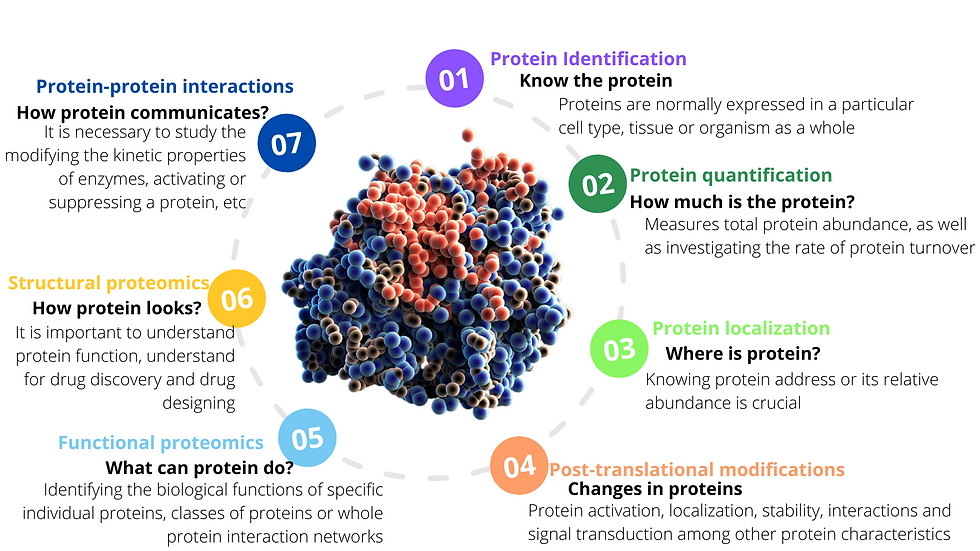
Studying proteins have been a challenge since start of the research world. There are analytical techniques which are divided as per the application of the study. There are low throughput techniques which can be multiplexed but make the range of proteins short for the study. It requires prior knowledge of proteins and have a long pre-processing protocol. There are other techniques for high throughput which allow wider range of samples to be analysed. These high throughput techniques have evolved in technologies over the period of 10 years and today it offers targeted and untargeted proteins study platforms.
Current MS techniques can be broadly divided into data-dependent and data-independent methods. In data-dependent methods, the selection of peptide fragments to be for subsequent MS/MS analysis depends on pre-established criteria. Whereas, in data-independent methods, all peptides (within a pre-determined mass range) are fragmented indiscriminately. Data-independent methods are relatively new but hold great promise for the future of quantitative proteomics. The advantages of this method are that it potentially can analyse all species that are present in a specific sample at detectable concentrations in a single assay and that the data can be retrospectively used as improved software and in silico databases become available.
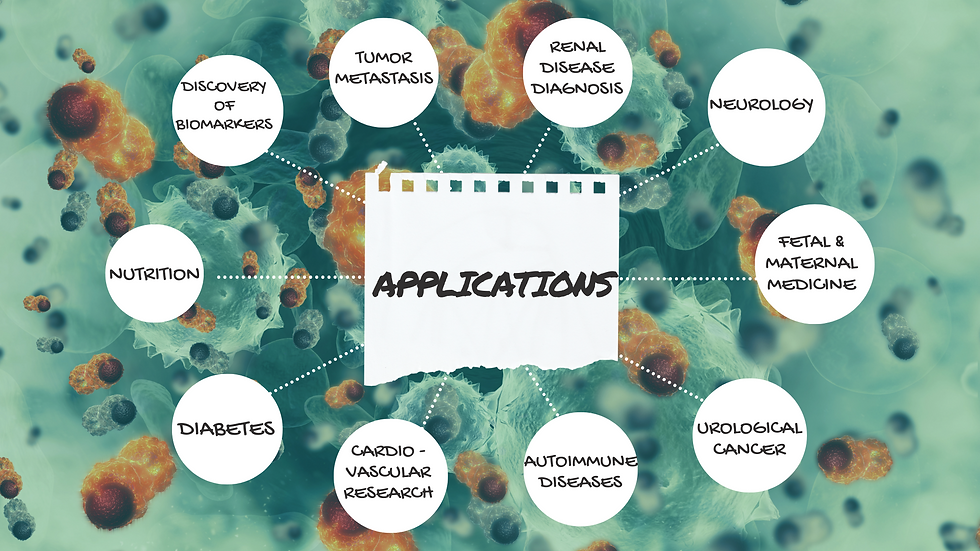
Applications of proteomics
-
For discovery of protein biomarkers
Biomarkers of drug efficacy and toxicity are becoming a key need in the drug development process. Mass spectral-based proteomic technologies are ideally suited for the discovery of protein biomarkers in the absence of any prior knowledge of quantitative changes
in protein levels. The success of any biomarker discovery effort will depend upon the quality of samples analysed, the ability to generate quantitative information on relative protein levels and the ability to readily interpret the data generated.
-
Study of Tumor Metastasis
Tumor metastasis is the dominant cause of death in cancer patients. However, the molecular and cellular mechanisms underlying tumor metastasis are still elusive. The identification of protein molecules with their expressions correlated to the metastatic process would
help to understand the metastatic mechanisms and thus facilitate the development of strategies for the therapeutic interventions and clinical management of cancer. Proteomic technology has been widely used in biomarker discovery and pathogenetic studies
including tumor metastasis.
-
In renal disease diagnosis
In the diagnosis and treatment of kidney disease, a major priority is the identification of disease-associated biomarkers. Its high-throughput and unbiased approach to the analysis of variations in protein expression patterns (actual phenotypic expression of genetic variation), promises to be the most suitable platform for biomarker discovery. Issues related to three such critical developmental tasks as follows: (i) completely defining the proteome in the various biological compartments (e.g. tissues, serum and urine) in both health and disease, which presents a major challenge given the dynamic range and complexity of such proteomes
(ii) achieving the routine ability to accurately and reproducibly quantify proteomic
expression profiles; and
(iii) developing diagnostic platforms that are readily applicable and technically feasible for use in the clinical setting that depend on the fruits of the preceding two tasks to profile multiple disease biomarkers.
-
In neurology
In neurology and neuroscience, many applications of proteomics have involved neurotoxicology and neurometabolism, as well as in the determination of specific proteomic aspects of individual brain areas and body fluids in neurodegeneration. Investigation of brain protein groups in neurodegeneration, such as enzymes, cytoskeleton proteins, chaperones, synaptosomal proteins and antioxidant proteins, is in progress as phenotype-related proteomics. An additional advantage is that hitherto unknown proteins have been identified as brain proteins.
-
In fetal and maternal medicine
Proteomics includes the characterization and functional analysis of all proteins that are expressed by the genome at a certain moment, under certain conditions. Since expression levels of many proteins strongly depend on complex, but well-balanced regulatory systems, the proteome, unlike the genome, is highly dynamic. This variation depends on the biological function of a cell, but also on signals from its environment. In (bio) medical research it has become increasingly apparent that cellular processes, in particular in disease, are determined by multiple proteins. Hence it is important not to focus on one single gene product (one protein), but to study the complete set of gene products (the proteome). In this way the multi-factorial relations underlying certain diseases may be unravelled potentially identifying therapeutical targets. For many diseases characterization of the functional proteome is crucial for elucidating alterations in protein expression and modifications. When proteins undergo non-genetically determined alterations such as alternative splicing, or post-translational modifications, e.g. phosphorylation or glycosylation, it may affect their function. Although abnormalities in splicing or post-translational modifications can cause a disease process, they can also be a consequence. An example is that patients with diabetes have high blood glucose which glycosylates hundreds or even thousands of proteins, including HbA1c which is used to monitor diabetic control.
-
Urological Cancer Research
Proteomic analysis allows the comparison of the proteins present in a diseased tissue sample with the proteins present in a normal tissue sample. Any proteins, which have been altered either quantitatively or qualitatively between the normal and diseased sample are likely to be associated with the disease process. These proteins can be identified and may be useful as diagnostic markers for the early detection of the disease or prognostic markers to predict the outcome of the disease or they may be used as drug targets for the development of new therapeutic agents.
-
In Autoantibody profiling for the study and treatment of autoimmune disease
Proteomics technologies enable the profiling of autoantibody responses using biological fluids derived from patients with autoimmune disease. They provide a powerful tool to characterize autoreactive B-cell responses in diseases including rheumatoid arthritis, multiple sclerosis, autoimmune diabetes, and systemic lupus erythematosus. Autoantibody profiling may serve purposes including classification of individual patients and subsets of patients based on their autoantibody fingerprint, examination of epitope spreading and antibody isotype usage, discovery and characterization of candidate autoantigens, and tailoring antigen-specific therapy. In the coming decades, proteomics technologies will broaden our understanding of the underlying mechanisms of and will further our ability to diagnose, prognosticate and treat autoimmune disease.
-
Cardiovascular research
The development of proteomics is a timely one for cardiovascular research. Analyses at the organ, sub cellular, and molecular levels have revealed dynamic, complex, and subtle intracellular processes associated with heart and vascular disease. The power and flexibility of proteomic analyses, which facilitate protein separation, identification, and characterization, should hasten our understanding of these processes at the protein level. Evolution of proteomic techniques has permitted more thorough investigation into molecular mechanisms underlying cardiovascular disease, facilitating identification not only of modified proteins but also of the nature of their modification. Continued development should lead to functional proteomic studies, in which identification of protein modification, in conjunction with functional data from established biochemical and physiological methods, has the ability to further our understanding of the interplay between proteome change and cardiovascular disease.
-
Diabetes research
The techniques involved in proteomics allow the global screening of complex samples of proteins and provide qualitative and quantitative
evidence of altered protein expression. This lends itself to the investigation of the molecular mechanisms underpinning disease processes and the
effects of treatment.
-
Nutrition Research
Proteomics holds great promise for discoveries in nutrition research, including profiles and characteristics of dietary and body proteins; digestion, absorption, and metabolism of nutrients; functions of nutrients and other dietary factors in growth, reproduction, and health;
biomarkers of the nutritional status and disease; and individualized requirements of nutrients. The proteome analysis is expected to play an important role in solving major nutrition-associated problems in humans and animals, such as obesity, diabetes, cardiovascular disease, cancer, aging, and intrauterine fetal retardation.
-
Future Prospective
Proteomic techniques document the expression of molecules which directly influence cell phenotype, and therefore provide data of clinical relevance. These techniques can be applied to a wide variety of organisms and biological samples, and can be applied to investigate protein expression in vitro or in vivo. Proteomic tools permit analysis of thousands of proteins and their PTMs and enables non-biased appraisals of the molecular biology of disease states, highlighting molecules which may be overlooked in hypothesis-driven scenarios. As described above, proteomic science is a relatively recent development. Currently, proteomics receives many criticisms, notably low reproducibility evidenced by gel-to-gel and inter-laboratory variation. Despite this, it is likely that these issues will diminish as protocols and reporting requirements become standardized and are universally applied. The investigative power of proteomics is greatly magnified when combined with other post-genomic techniques. Combined proteomic and metabolomic analyses provide direct evidence of the effect of protein expression on cellular processes.
In discovery proteomics, proteome analysis can be performed in two different strategies, bottom-up and top-down approaches, respectively. In the bottom-up approach, a crude protein mixture undergoes protease digestion first, and then separation by liquid chromatography, followed by MS analysis. In the top-down method, proteins are characterized by MS without prior proteolysis. This type of approach has the advantage of providing greater sequence coverage, resolution of sequence ambiguities and preservation of PTMs.

